
有源医疗器械
2021-03-25
海外认证
Overseas Certification
经典案例
Classic Case
II类产品占FDA医疗器械产品目录的46%左右,实行的是特殊控制(Special Control)①,除进行企业注册和产品列名外,还需实施GMP规范和递交510(k)申请 【极少部分产品豁免提交510(k)】。
①特殊控制包括以下内容:上市前监督研究、跟踪随访使用产品的患者、使用指南(例如:防护服的使用方法)、产品性能标准、建议及其他措施、特殊标识(例如:882.5970 头部矫形器)
510(k)文件是向FDA递交的上市前申请文件,目的是证明申请上市的器械与不受上市前批准(PMA)影响的合法上市器械同样安全有效,即实质性等效(substantial equivalence)②。申请者必须把申请上市的器械与现在美国已获批的一种或多种相似器械(predicate device)进行对比,得出并且支持等价器械的结论。
②实质性等效是指,与合法销售的器械相比,该器械:
➣ 具有相同的预期用途,而且
➣ 与已获批准的类似器械具有相同的技术特点;
或者:
➣ 具有相同的预期用途,虽然
➣ 具有不同的技术特点,但510(k)中提供的信息:
i.未引发新的安全性和有效性问题,而且
ii.证明与已获批准的类似器械相比,具有相当的安全性和有效性
➣ 提交510(k)文件的3种类型:
1.传统510(k):可以被用于任何适用器械的首次510(k)提交,以此证明申请上市的器械与predicate devices 是实质性等效的,其中包含21 CFR第807.87部分中所列的所有要素;在90天内审核完毕;
2.特殊510(k):适用于已获批器械的设计或标签变更(包括某些适应症的变更),申请人按照21 CFR 第820.30部分(设计控制)对变更进行评估,同时一并递交符合设计控制原则的声明;在30天内审核完毕;
3.简略510(k):如果所提交的器械类型有特定的公认标准,或者FDA已经发布了关于该器械分类界定的指导文件,那么建议采用简略510(k) 提交;在60天内审核完毕。
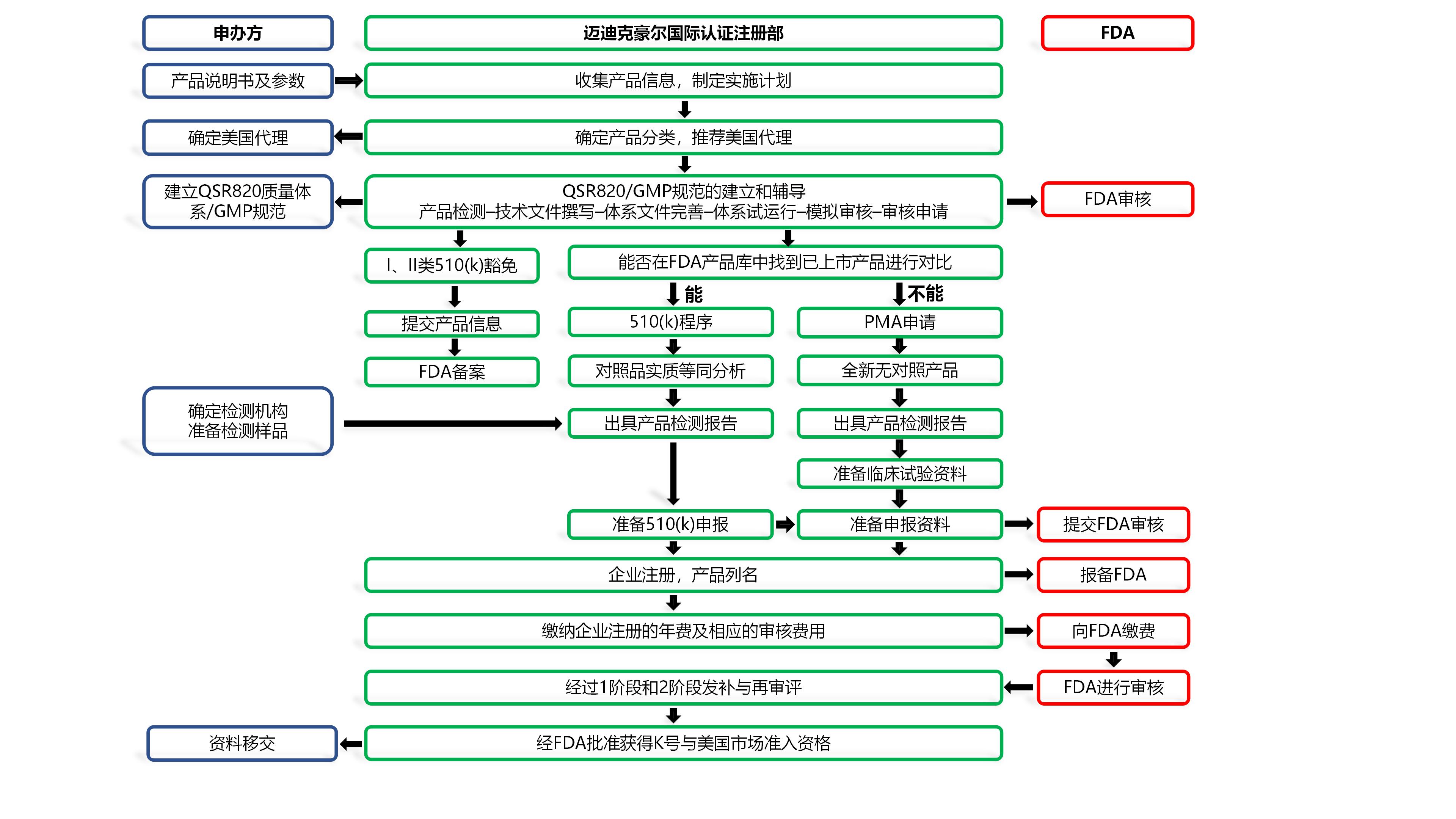
➣ CRCS的服务流程
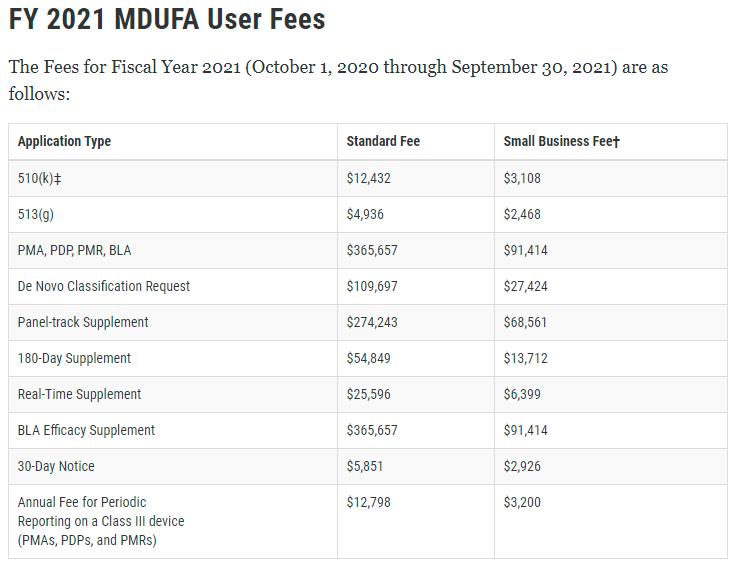
➣ FDA行政费:
➣ FDA的审查流程与预估的时间线

扫二维码用手机看

- 微信公众号 -
联系方式
北京迈迪克豪尔医药技术咨询服务有限公司